|
ORENITRAM (treprostinil) extended-release tablets
|
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ORENITRAM ® safely and effectively. See Full Prescribing Information for ORENITRAM.
ORENITRAM (treprostinil) extended-release tablets, for oral use
Initial U.S. Approval: 2002
INDICATIONS AND USAGE
Orenitram is a prostacyclin vasodilator indicated for:
Treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise capacity. The study that established effectiveness included predominately patients with WHO functional class II-III symptoms and etiologies of idiopathic or heritable PAH (75%) or PAH associated with connective tissue disease (19%). (1.1)
As the sole vasodilator, the effect on exercise is small. Orenitram has not been shown to add to other vasodilator therapy. (1.1)
DOSAGE AND ADMINISTRATION
Give with food. Swallow tablets whole; use only intact tablets. (2.1)
Starting dose: 0.25 mg BID or 0.125 mg TID. (2.1)
Titrate by 0.25 mg or 0.5 mg BID or 0.125 mg TID, not more than every 3 to 4 days as tolerated. (2.1)
Maximum dose is determined by tolerability. (2.1)
Mild hepatic impairment (Child Pugh Class A): Initiate at 0.125 mg BID. Increment at 0.125 mg BID every 3 to 4 days. (2.2)
Avoid use in patients with moderate hepatic impairment. (2.2)
DOSAGE FORMS AND STRENGTHS
Extended-Release Tablets: 0.125 mg, 0.25 mg, 1 mg and 2.5 mg. (3)
CONTRAINDICATIONS
Severe hepatic impairment (Child Pugh Class C). (4)
WARNINGS AND PRECAUTIONS
Do not abruptly discontinue dosing. (2.4, 5.1)
Increased risk of bleeding, particularly in patients receiving anticoagulants. (5.2)
Do not take Orenitram with alcohol (5.3)
In patients with diverticulosis Orenitram tablets can become lodged in a diverticulum. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence >10%) reported in clinical studies with Orenitram are headache, nausea, and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact United Therapeutics Corp. at 1-866-458-6479 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Blood pressure lowering drugs (e.g., diuretics, antihypertensive agents, or vasodilators): Risk of hypotension (7.1)
When co-administered with strong CYP2C8 inhibitors the initial dose is 0.125 mg BID with 0.125 mg BID dose increments every 3 to 4 days. (2.3, 7.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2014
FULL PRESCRIBING INFORMATION: CONTENTS*
1. INDICATIONS AND USAGE
1.1 Pulmonary Arterial Hypertension
Orenitram is indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise capacity. The study that established effectiveness included predominately patients with WHO functional class II-III symptoms and etiologies of idiopathic or heritable PAH (75%) or PAH associated with connective tissue disease (19%).
When used as the sole vasodilator, the effect of Orenitram on exercise is about 10% of the deficit, and the effect, if any, on a background of another vasodilator is probably less than this. Orenitram is probably most useful to replace subcutaneous, intravenous, or inhaled treprostinil, but this use has not been studied.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
Individualize dosing of Orenitram according to clinical response.
Take Orenitram with food. Swallow Orenitram tablets whole; do not crush, split, or chew.
The recommended starting dose of Orenitram is 0.25 mg twice daily (BID) with food, taken approximately 12 hours apart or 0.125 mg three times daily (TID) with food, taken approximately 8 hours apart. Increase the dose as tolerated to achieve optimal clinical response. The recommended increment is 0.25 or 0.5 mg BID or 0.125 mg TID every 3-4 days. If dose increments are not tolerated consider titrating slower.
The maximum dose is determined by tolerability. The mean dose in a controlled clinical trial at 12 weeks was 3.4 mg BID. Maximum doses studied were 12 mg BID in the 12-week blinded study and up to 21 mg BID in an open-label long-term study.
If intolerable pharmacologic effects occur, decrease the dose in increments of 0.25 mg. Avoid abrupt discontinuation [see Warnings and Precautions (5.1)].
2.2 Hepatic Impairment
In patients with mild hepatic impairment (Child Pugh Class A) start at 0.125 mg BID with 0.125 mg BID dose increments every 3 to 4 days. Avoid use of Orenitram in patients with moderate hepatic impairment (Child Pugh Class B). Orenitram is contraindicated in patients with severe hepatic impairment (Child Pugh Class C) [see Contraindications (4), Warnings and Precautions (5.4), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
2.3 Concomitant Administration with CYP2C8 Inhibitors
When co-administered with strong CYP2C8 inhibitors (e.g., gemfibrozil) the initial dose is 0.125 mg BID with 0.125 mg BID dose increments every 3 to 4 days.
2.4 Interruptions and Discontinuation
If a dose of medication is missed, the patient should take the missed dose as soon as possible, with food. If a patient misses two or more doses, restart at a lower dose and re-titrate.
In the event of a planned short-term treatment interruption for patients unable to take oral medications, consider a temporary infusion of subcutaneous or intravenous treprostinil. To calculate the total daily dose (mg) of treprostinil for the parenteral route divide the oral total daily dose by 5.
When discontinuing Orenitram, reduce the dose in steps of 0.5 to 1 mg per day [see Warnings and Precautions (5.1)].
3 DOSAGE FORMS AND STRENGTHS
Orenitram (treprostinil extended-release) is available in the following four strengths:
- 0.125 mg [White tablet imprinted with UT 0.125]
- 0.25 mg [Green tablet imprinted with UT 0.25]
- 1 mg [Yellow tablet imprinted with UT 1]
- 2.5 mg [Pink tablet imprinted with UT 2.5]
4 CONTRAINDICATIONS
Severe hepatic impairment (Child Pugh Class C) [see Use In Specific Populations (8.6) and Clinical Pharmacology (12.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Worsening PAH Symptoms upon Abrupt Withdrawal
Abrupt discontinuation or sudden large reductions in dosage of Orenitram may result in worsening of PAH symptoms.
5.2 Risk of Bleeding
Orenitram inhibits platelet aggregation and increases the risk of bleeding.
5.3 Increased Exposure with Alcohol
Do not take Orenitram with alcohol as release of treprostinil from the tablet may occur at a faster rate than intended.
5.4 Use in Patients with Blind-end Pouches
The tablet shell does not dissolve. In patients with diverticulosis, Orenitram tablets can lodge in a diverticulum.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In a 12-week placebo-controlled monotherapy study (Study 1; WHO Group 1; functional class II-III), the most commonly reported adverse reactions that occurred in patients receiving Orenitram included: headache, nausea, and diarrhea. Table 1 lists the adverse reactions that occurred at a rate on Orenitram at least 5% higher than on placebo.
Orenitram patients in Table 1 for Study 1 (N = 151) had access to 0.25 mg tablets at randomization. Approximately 91% of such patients experienced an adverse reaction, but only 4% discontinued therapy for an adverse reaction (compared to 3% receiving placebo). The overall discontinuation rate for any reason was 17% for active and 14% for placebo.
Table 1: Adverse Reactions with Rates at Least 5% Higher on Orenitram Monotherapy than on Placebo
|
Reaction |
Orenitram
N=151 |
Placebo
N=77 |
|
Headache |
63% |
19% |
|
Diarrhea |
30% |
16% |
|
Nausea |
30% |
18% |
|
Flushing |
15% |
6% |
|
Pain in jaw |
11% |
4% |
|
Pain in extremity |
14% |
8% |
|
Hypokalemia |
9% |
3% |
|
Abdominal discomfort |
6% |
0% |
Orenitram was studied in a long-term, open-label extension study in which 824 patients were dosed for a mean duration of approximately 2 years. About 70% of patients continued treatment with Orenitram for at least a year. The mean dose was 4.2 mg BID at one year. The adverse reactions were similar to those observed in the placebo-controlled trials.
7 DRUG INTERACTIONS
7.1 Antihypertensive Agents or Other Vasodilators
Concomitant administration of Orenitram with diuretics, antihypertensive agents or other vasodilators increases the risk of symptomatic hypotension.
7.2 Anticoagulants
Treprostinil inhibits platelet aggregation; there is increased risk of bleeding, particularly among patients receiving anticoagulants.
7.3 Effect of CYP2C8 Inhibitors
Co-administration of Orenitram and the CYP2C8 enzyme inhibitor gemfibrozil in healthy adult volunteers increases exposure to treprostinil. Reduce the starting dose of Orenitram to 0.125 mg BID and use 0.125 mg BID increments every 3 to 4 days [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C.
Animal reproductive studies with treprostinil diolamine have shown an adverse effect on the fetus. There are no adequate and well-controlled studies in humans.
In rats, treatment with treprostinil diolamine had no effect on reproductive performance or sperm motility at doses up to 10 mg/kg/day. The exposures at this dose level are about 10- (male) to 18- (female) fold the usual human exposure at the mean dose of 3.4 mg BID.
In pregnant rats, reversible, dose-dependent decreases in body weight gain and food consumption were observed during the first four days of dosing in animals administered 10, 20 and 30 mg/kg/day treprostinil diolamine. In a dose range-finding study, there was a 17% decrease in the pregnancy rate in the animals administered 20 and 30 mg/kg/day. One dam in each of the 20 and 30 mg/kg/day had litters with no viable fetuses. In the definitive study (0, 5, 10 and 20 mg/kg/day), there were four treatment-related deaths, and a 32% decrease in the pregnancy rate for rats administered 20 mg/kg/day. There was an 8% decrease in the pregnancy rate in the animals administered 10 mg/kg/day. Across both studies, an increase in post-implantation loss was observed in animals administered 10 to 30 mg/kg/day, and a significant decrease in the mean number of live births was seen at dose levels ≥10 mg/kg/day. The no observed adverse effect level was 5 mg/kg/day (maternal, fetal viability and growth), and 20 mg/kg/day (teratogenicity), the highest dose tested in the definitive study. The exposures at 5 and 20 mg/kg/day doses represent 13 and 55 times, respectively, the human exposure.
For F1 progeny, a decreased copulation index was observed at the 5 and 10 mg/kg/day treprostinil diolamine dose levels in rats. The no observed effect levels for physical development, reflex development, exploratory behavior, learning and memory, and sexual maturation was 10 mg/kg/day. The no observed effect level for F1 progeny general development (based on body weight) was 10 mg/kg/day for females and ≤ 2.5 mg/kg/day for males; the no observed effect level for F1 reproductive performance was 2.5 mg/kg/day or 6 times the human exposure.
In pregnant rabbits, the primary maternal adverse effects were gastrointestinal disturbance; dose-dependent decreases in mean body weight, body weight gain, and food consumption were observed. During the post-dose phase, the effect was reversed. In a dose range-finding study, there was a 17% decrease in the pregnancy rate for animals administered 4 mg/kg/day. A dose-dependent increase in post-implantation loss was observed. Two dams administered 4 mg/kg/day had litters with no viable fetuses; the mean fetal weight was slightly decreased in animals administered 4 mg/kg/day. In the definitive study, mean fetal weights were significantly decreased in animals administered 0.5 to 3 mg/kg/day of treprostinil diolamine. At doses of 1.5 and 3 mg/kg/day, external fetal and soft tissue malformations were observed in a few fetuses, and the total fetal skeletal malformations were significantly increased. The no observed adverse effect level was less than 0.5 mg/kg/day (maternal), 1.5 mg/kg/day (fetal viability and growth), and 0.5 mg/kg/day (teratogenicity). The 0.5 mg/kg/day dose represents about 5 times the human exposure.
8.2 Labor and Delivery
The effect of Orenitram on labor and delivery in humans is unknown. No treprostinil treatment-related effects on labor and delivery were seen in animal studies.
8.3 Nursing Mothers
It is not known whether treprostinil is excreted in human milk or absorbed systemically after ingestion. Because many drugs are excreted in human milk, choose Orenitram or breastfeeding.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of Orenitram did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic or cardiac function, and of concomitant disease or other drug therapy.
8.6 Patients with Hepatic Impairment
There is a marked increase in the systemic exposure to treprostinil in hepatically impaired patients [see Dosage and Administration (2.2), Contraindications (4), and Clinical Pharmacology (12.3)].
8.7 Patients with Renal Impairment
No dose adjustments are required in patients with renal impairment. Orenitram is not removed by dialysis [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Signs and symptoms of overdose with Orenitram during clinical trials reflect its dose-limiting pharmacologic effects and include severe headache, nausea, vomiting, diarrhea, and hypotension. Treat supportively.
11 DESCRIPTION
Orenitram is an extended release osmotic tablet for oral administration. Orenitram is formulated as the diolamine salt of treprostinil, a tricyclic benzindene analogue of prostacyclin. The chemical name is Acetic acid, 2-[[(1R,2R,3aS,9aS)-2,3,3a,4,9,9a-hexahydro-2-hydroxy-1-[(3S)-3-hydroxyoctyl]-1H-benz[f]inden-5-yl]oxy]-, complexed with 2,2'-iminobis[ethanol] (1:1). The molecular formula is C23H34O5.C4H11NO2, the molecular weight is 495.65, and it has the following structural formula:
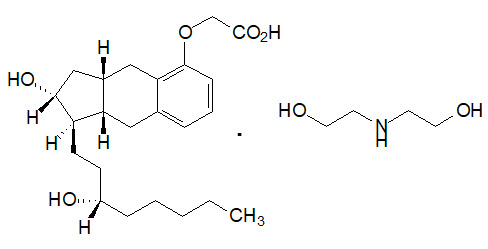
Orenitram tablets are formulated in four strengths, which contain 0.125 mg of treprostinil (equivalent to 0.159 mg treprostinil diolamine), 0.25 mg of treprostinil (equivalent to 0.317 mg treprostinil diolamine), 1 mg of treprostinil (equivalent to 1.27 mg treprostinil diolamine), or 2.5 mg of treprostinil (equivalent to 3.17 mg treprostinil diolamine). The formulations also contain xylitol, maltodextrin, sodium lauryl sulfate, magnesium stearate, cellulose acetate, triethyl citrate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, and talc. In addition tablets may contain colorants FD&C Blue #2, iron oxide yellow, and iron oxide red. The imprinting ink contains shellac glaze, ethanol, isopropyl alcohol USP, iron oxide black, n-butyl alcohol, propylene glycol, and ammonium hydroxide.
Orenitram is designed to release treprostinil at a near zero-order rate using an osmotic tablet technology. The tablet core is coated with a semi-permeable membrane and has a laser-drilled aperture through the membrane. Upon contact with water (e.g., after ingestion), the core tablet absorbs water through the semi-permeable membrane. The water dissolves the water-soluble treprostinil diolamine and the water-soluble osmotic excipients, which creates hydrostatic pressure within the membrane, eventually forcing the drug out through the tablet at a controlled rate.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of ActionThe major pharmacologic actions of treprostinil are direct vasodilation of pulmonary and systemic arterial vascular beds, inhibition of platelet aggregation, and inhibition of smooth muscle cell proliferation.
12.2 Pharmacodynamics
In a clinical trial of 240 healthy adult volunteers, single doses of inhaled treprostinil 54 µg (the target clinical dose) and 84 µg (supratherapeutic inhalation dose) prolonged the corrected QTc interval by approximately 10 msec. The QTc effect dissipated rapidly as the concentration of treprostinil decreased. Orenitram has not been eva luated in a thorough QTc study.
12.3 Pharmacokinetics
In patients with PAH, pharmacokinetics of treprostinil is dose-proportional for systemic exposure (AUC0-t) over the dose range of 0.5 and 15 mg BID. Upon repeat administration with a BID regimen, the accumulation in the systemic exposures to treprostinil is minimal and results in a peak-to-trough ratio of approximately 7. However, a TID regimen will reduce the peak-to-trough fluctuations to approximately 2.5 for the same total daily dose.
Absorption
The absolute oral bioavailability of Orenitram is approximately 17%. Maximum treprostinil concentrations occur between approximately 4 and 6 hours following Orenitram administration.
The absorption of Orenitram is affected by food. The AUCinf of treprostinil was increased by 49% and the Cmax was increased by an average of 13% when Orenitram was administered following a high-fat, high-calorie meal compared to fasting conditions in healthy volunteers. The relative bioavailability of treprostinil following oral administration of Orenitram 1 mg is not significantly altered by meal types ranging from 250 to 500 calories in healthy volunteers.
Distribution
The treprostinil component of Orenitram is highly bound to human plasma proteins, approximately 96% over a treprostinil concentration range of 0.01-10 µg/mL.
Metabolism and Excretion
In a study conducted in healthy volunteers using [14C] treprostinil, treprostinil was extensively metabolized on the side chain of the molecule via oxidation, oxidative cleavage, dehydration, and glucuronic acid conjugation. Treprostinil is primarily metabolized by CYP2C8 and to a lesser extent by CYP2C9. No new metabolites are found upon oral administration compared to parenteral administration of treprostinil. Only 1.13% and 0.19% is excreted as unchanged parent drug in the feces and urine, respectively. Based on in vitro studies treprostinil does not inhibit or induce major CYP enzymes [see Drug Interactions (7.3)].
Special Populations
Hepatic Impairment: In subjects with mild (n=8) hepatic impairment, administration of a single 1 mg dose of Orenitram resulted in a mean Cmax and an AUC0-inf that were 1.6- and 2.1-fold, respectively values seen in healthy subjects. With moderate impairment (n=8), the corresponding ratios were 4.0- and 4.8-fold, and with severe impairment (n=6), they were 4.8- and 7.6-fold [see Dosage and Administration (2.2), Contraindications (4), and Use in Specific Populations (8.6)].
Renal Impairment: In patients with severe renal impairment requiring dialysis (n=8), administration of a single 1 mg dose of Orenitram pre- and post-dialysis resulted in an AUC 0-inf that was not significantly altered compared to healthy subjects.
Drug Interactions
Results of drug interaction studies are shown in Figure 1. Only for the strong CYP2C8 inhibitor does the interaction affect dosing.
Figure 1: Impact of Co-Administered Drugs on the Systemic Exposure of Treprostinil 1 mg Compared to Orenitram Administered Alone

Warfarin: A drug interaction study was carried out with Remodulin co-administered with warfarin (25 mg/day) in healthy volunteers. There was no clinically significant effect of either medication on the pharmacokinetics of treprostinil. Additionally, treprostinil did not affect the pharmacokinetics or pharmacodynamics of warfarin. The pharmacokinetics of R- and S- warfarin and the international normalized ratio (INR) in healthy subjects given a single 25 mg dose of warfarin were unaffected by continuous subcutaneous infusion of treprostinil at an infusion rate of 10 ng/kg/min.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Oral administration of treprostinil diolamine to Tg.rasH2 mice at 0, 5, 10 and 20 mg/kg/day in males and 0, 3, 7.5 and 15 mg/kg/day in females daily for 26 weeks did not significantly increase the incidence of tumors. The exposures obtained at the highest dose levels used in males and females are about 8- and 17-fold, respectively, the human exposure at the mean dose of 3.4 mg BID.
In vitro genotoxicity studies with high doses of treprostinil did not demonstrate any mutagenic or clastogenic effects. Treprostinil diolamine was tested in vivo in a rat micronucleus assay and did not induce an increased incidence of micronucleated polychromatic erythrocytes.
No adverse effect doses for fertility, fetal viability / growth, fetal development (teratogenicity), and postnatal development were determined in rats. In pregnant rabbits, external fetal and soft tissue malformations and fetal skeletal malformation occurred with the no observed adverse effect level for these adverse effects of 0.5 mg/kg/day (5 times the human exposure) [see Use in Specific Populations (8.1)].
14 CLINICAL STUDIES
14.1 Clinical Trials in Pulmonary Arterial Hypertension (PAH)
Three multi-center, randomized, double-blind studies were conducted and compared Orenitram to placebo in a total of 349 (Study 1), 350 (Study 2), and 310 (Study 3) patients with PAH.
Study 1 (effect seen with no background vasodilator)
Study 1 was a 12-week, randomized (2:1 Orenitram to placebo), double-blind, placebo-controlled, international efficacy and safety study of Orenitram in patients with WHO Group 1 PAH not currently receiving PAH therapy. The primary efficacy endpoint was placebo-corrected change in six-minute walk distance (6MWD) from Baseline to Week 12. Study drug dose wa |
|

